Der große Erfolg bei der Suche nach Krankheitsverursachenden Genen in komplexen Erkrankungen stellte sich jedoch erst ein, als es gelang, mit genomweiten Assoziationsstudien (GWA), mehrere Genvarianten die gehäuft bei KHK, Hypercholesterinämie oder Diabetes mellitus auftreten, eindeutig zu identifizieren.
Genomweite Assoziationsstudien die Innovation
Auch mittels genomweiter Assoziationsstudien konnte unsere Arbeitsgruppe für die Herzinfarktgenetik einen ganz entscheidenden Beitrag zur Identifizierung neuer Genvarianten leisten. Gemeinsam mit deutschen (Universität zu Regensburg und Helmholtz-Zentrum, München), britischen und französischen Kollegen analysierten wir die genetischen Daten von knapp 3.000 Patienten und 4.500 gesunden Probanden aus zwei unabhängigen genomweiten Assoziations- studien zum Herzinfarkt. Dabei ermittelten wir insgesamt sieben Regionen auf unterschiedlichen Chromosomen, die allein oder im Zusammenspiel mit anderen, das Risiko für koronare Herzerkrankungen und Herzinfarkt deutlich erhöhen können. Der Hauptlokus auf Chromosom 9p21.3, der durch diese Studie identifiziert wurde, verdoppelt für homozygote Träger des Risiko-Allels das Erkrankungsrisiko. Der Einfluss dieses Genlokus konnte auch in weiteren GWAs und Replikationsstudien zum Herzinfarkt bestätigt werden. Damit ist die Region auf Chromosom 9p21.3 die bislang am sichersten mit dem genetischen Risiko für KHK oder Herzinfarkt assoziierte Genregion. In dieser Region liegen zwei protein-kodierende Gene (p16INK4a und p15INK4b), die eine Rolle bei der Regulation des Zellwachstums spielen, sowie ein RNA-kodierendes Gen (ANRIL). Bislang ist aber noch nicht nachgewiesen worden, welches der o.g. Gene tatsächlich krankheits-verursachend ist.
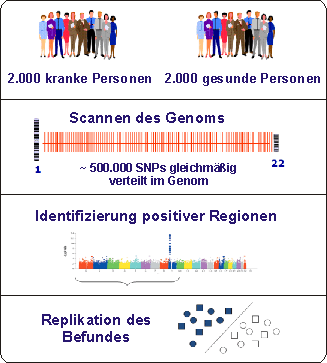
Genomweite Analysen benötigen keine hypothetischen Annahmen bezüglich der Pathophysiologie der Erkrankung. Durch die Analyse von genetischen Markern, die engmaschig das gesamte Erbgut überspannen, können genetische Abweichungen erkannt und somit Regionen identifiziert werden, in denen ein krankheitsverursachendes Gen mit hoher Wahrscheinlichkeit lokalisiert ist. Mittels dieser Methodik können mehr als 500.000, mittlerweile sogar 1.000.000, genetischer Varianten (sog. SNPs, Einzelbasenpaar- Austausche) in Tausenden von Patienten simultan analysiert werden, was als klarer Vorteil gegenüber anderen Analyseverfahren anzusehen ist. Die Umsetzung dieser methodischen Innovation hat in den vergangenen Monaten zu einer explosionsartigen Identifikation etlicher bislang unbekannter Gene bzw. Genregionen für verschiedene komplexe Erkrankungen geführt, wie z.B. Diabetes, Bluthochdruck, Adipositas, Restless Legs Syndrom und der koronaren Herzerkrankung geführt.